IB20047
Inteligencia Artificial de Altas Prestaciones para optimizar el diseño de secuencias de ARN

El diseño de secuencias de ARN consiste en, dada una estructura secundaria, encontrar una secuencia de bases (secuencia de ARN) que se pliegue de forma única en esta estructura o informar que dicha secuencia no existe. Este problema de diseñar secuencias de ARN que se adapten a una estructura secundaria dada, se conoce también como plegamiento inverso de ARN.
La motivación para estudiar este problema proviene de la enorme importancia de las funciones que realizan determinadas secuencias de ARN, las cuales están fuertemente influenciadas por las estructuras secundarias en las que se pliegan. Por lo tanto, la utilización de técnicas computacionales que permitan diseñar secuencias de ARN con una función específica es crucial, pues ahorra muchas horas de laboratorio. Por otro lado, el diseño de secuencias de ARN tiene una gran relevancia en diferentes áreas, como la investigación farmacéutica, la bioquímica, la biología sintética o las nano-estructuras de ARN. Estas dos últimas áreas apuntan a crear secuencias de ARN mejorado, es decir, secuencias de ARN con propiedades deseables. Por lo tanto, hay muchos algoritmos y productos de software que resuelven el problema del plegamiento inverso de ARN, tales como RNAinverse, RNA-SSD, MODENA, antaRNA, NUPACK, fRNAkenstein, etc. Desafortunadamente, la gran mayoría son heurísticas clásicas que tienen un tiempo de ejecución exponencial.
El objetivo de este proyecto es crear, mejorar y difundir nuevas y avanzadas técnicas de inteligencia artificial capaces de optimizar el diseño de secuencias de ARN. Además, queremos avanzar en el uso de tecnologías de computación de altas prestaciones de forma que podamos reducir los tiempos de ejecución a la hora de diseñar secuencias de ARN complejas. Por último, este proyecto tiene transferencias previsibles a la industria dado que todas las técnicas desarrolladas pueden aplicarse a otros dominios, tanto del sector biotecnológico como de otros sectores de interés para la comunidad autónoma de Extremadura, p. ej. agroalimentario, energético o turístico.

Junto al ácido desoxirribonucleico (ADN) y las proteínas, el ácido ribonucleico (ARN) es una molécula que desempeña una función muy importante. El ARN es una cadena de nucleótidos (o bases) y puede representarse como una secuencia en un alfabeto de cuatro letras, correspondientes a las bases nitrogenadas: Adenina (A), Uracilo (U), Citosina (C) y Guanina (G). A diferencia del ADN, el ARN es monocatenario y se repliega sobre sí mismo: algunas de las bases están unidas entre sí (emparejadas) para formar una estructura estable y compacta. Este emparejamiento forma lo que se conoce como la estructura secundaria de la molécula de ARN. Por otro lado, la estructura primaria es la secuencia de nucleótidos y la estructura terciaria es la forma 3D. Predecir cómo se pliega una molécula de ARN es vital para comprender su función biológica [BRS20].
La biología sintética es una disciplina emergente con ramificaciones que van desde la detección de moléculas dentro de la célula a la creación de genomas sintéticos y nuevas formas de vida [GSA+16]. Grupos pioneros en este campo han obtenido resultados espectaculares, p. ej. la síntesis combinatoria de redes de genes, la síntesis de genomas o la reacción de hibridación en cadena. La mayor parte de los trabajos en el campo de la biología sintética se han concentrado en lo que se puede considerar como genómica sintética, p. ej. la regulación sintética de genes o el desarrollo de bloques genéticos.
Como vimos anteriormente, la estructura de ARN determina en gran medida su función. Dada la gran cantidad de nuevos tipos de ARN con funciones complejas, y dada la actual variedad de algoritmos para predicción de estructura de ARN [GSA+16], de búsqueda de motifs estructurales [AHC07], etc.; está cada vez más claro que uno de los avances más importantes en la biología sintética trataría el diseño y validación experimental de nuevas estructuras sintéticas de ARN.
Dada una estructura secundaria de ARN cualquiera (también llamada estructura objetivo), se debe encontrar una secuencia de pares de bases nitrogenadas (A, U, C, G) cuyo plegamiento corresponda con esta estructura. Este problema se conoce como el plegamiento inverso de ARN (RNA inverse folding, en inglés) o diseño de secuencias de ARN (Designing RNA sequences, en inglés). Teniendo en cuenta que la estructura secundaria de ARN es una parte esencial de la estructura terciaria, y el plegamiento tiene lugar de forma jerárquica, cualquier herramienta capaz de resolver el problema del plegamiento inverso para estructura secundaria es, claramente, un gran paso hacia el diseño de ARN funcional.
Este problema se puede resolver mediante búsqueda por fuerza bruta. Desafortunadamente, su complejidad crece exponencialmente, de forma que para una estructura objetivo S de longitud n, existe un total de 4n posibles secuencias. Este límite superior puede refinarse si tenemos en cuenta que las posiciones emparejadas deben formar pares de bases válidos, como: AU, UA, GC, CG, GU o UG. Por lo tanto, dada una estructura objetivo (S) con u nucleótidos no emparejados y p emparejados, el número de secuencias válidas será igual a 6p/24u.
Una notación muy utilizada para representar la estructura secundaria es la notación punto-paréntesis (dot-bracket notation, en inglés), donde cada carácter representa una base, en caso de los paréntesis indican que emparejamiento con otra base posterior. El número de paréntesis de apertura y cierre debe ser siempre el mismo. Veamos un ejemplo de una estructura objetivo (S) en notación punto-paréntesis:
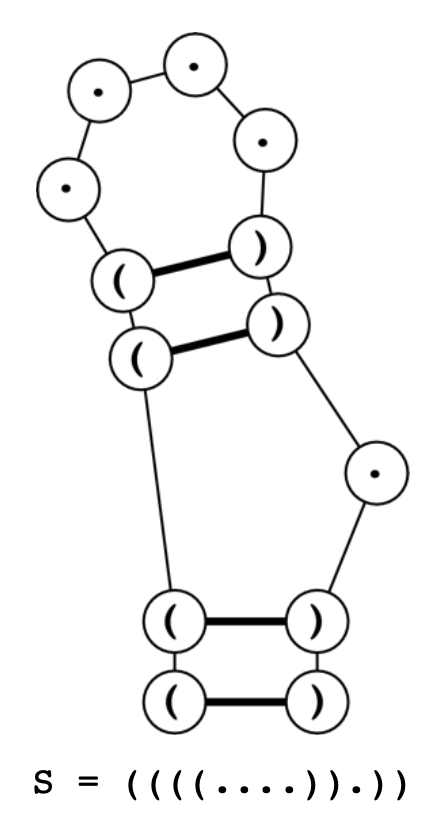
La longitud de la secuencia S es de 13 nucleótidos y su composición incluye 8 nucleótidos emparejados (p=8) y 5 nucleótidos no emparejados (u=5). Para esta pequeña estructura, el número de secuencias de ARN candidatas es superior al millón (68/245), concretamente, 1.327.104 posibles secuencias.
A continuación se muestran tres posibles secuencias de ARN (secuencias candidatas):
x1 = GGACUACGGUACC
x2 = GGCCUGCGGGACC
x3 = ACCCGAGAGGUGA
Para poder evaluar las tres posibilidades (x1, x2, x3), debemos predecir su estructura secundaria, para ello podemos utilizar cualquier algoritmo de plegamiento (fold):

S = ((((....)).)) .
x1 = GGACUACGGUACC ((((....)).)) 100%
x2 = GGCCUGCGGGACC ((((....)).)) 100%
x3 = ACCCGAGAGGUGA ..((....))... 69,2%
Únicamente las secuencias candidatas x1 y x2 resuelven el problema de forma satisfactoria, ya que su estructura secundaria es idéntica (100% de similitud) a la estructura objetivo (S).
Observamos que pueden existir infinidad de secuencias que resuelvan el problema de plegamiento inverso, sin embargo, no todas tienen la misma relevancia a nivel biológico. Por ello, para imitar las características biológicas de las secuencias y estructuras de ARN, se deben considerar algunas restricciones de secuencia y energía a la hora de diseñar las secuencias de ARN [ZPV+13]. En las restricciones de secuencia, se pueden restringir las posiciones de secuencia a un nucleótido fijo o a un subconjunto de nucleótidos. En cuanto a la restricción de energía, se puede especificar un intervalo para los rangos de energía libre de las secuencias diseñadas. La mayoría de estas restricciones pueden ser consideradas como funciones objetivo a optimizar y contradictorios entre sí, convirtiendo el problema del plegamiento inverso en un problema de optimización multiobjetivo.
En la literatura se pueden encontrar diversas referencias sobre las últimas tendencias relacionadas con la inteligencia artificial y la computación de altas prestaciones en el ámbito de la biología computacional [CHF20], [Cec19]. Sin embargo, en esta subsección repasaremos únicamente los antecedentes y el estado del actual estrechamente relacionado con el problema biológico descrito en la subsección anterior: diseño de secuencias de ARN.
Varios autores han decidido dar solución al problema del plegamiento inverso de ARN (Diseño de ARN). La gran mayoría de ellos comienza con una secuencia inicial que se modifica iterativamente hasta que se pliega en la estructura objetivo o se alcanza algún criterio de parada.
El primer algoritmo desarrollado para el diseño de ARN fue RNAinverse [HFS+94]. Está contenido en la librería Vienna Package [GLB+08]. RNAinverse divide la estructura objetivo de entrada en estructuras más pequeñas. A continuación, mediante el uso de un procedimiento de caminata aleatoria adaptativa, intenta minimizar la distancia del par de bases.
En 2004, Andronescu et al. presentaron RNA Secondary Structure Designer (RNA-SSD) [AFH+04], un algoritmo diferente a RNAinverse y más eficiente para el problema de plegamiento inverso de ARN. De manera similar a RNAinverse, RNA-SSD utiliza un enfoque de divide y vencerás mediante la descomposición jerárquica de la estructura objetivo de entrada. El algoritmo RNA-SSD es capaz de resolver estructuras para las cuales RNAinverse no puede encontrar soluciones.
Busch y Backofen [BB06] desarrollaron un algoritmo para este problema, denominándolo IN-verse FOlding of RNA (INFO-RNA). Su enfoque (INFO-RNA) consta de dos pasos básicos, un método de programación dinámica para obtener secuencias iniciales de calidad y una búsqueda local estocástica. Los autores concluyen que INFO-RNA se comporta mejor que RNAinverse y, en la mayoría de los casos, mejor que RNA-SSD. La principal desventaja de INFO-RNA es que las secuencias de ARN devueltas presentan un alto contenido de GC y tienden a tener poca semejanza con el ARN biológicamente activo.
En [Tan10], A. Taneda aplica el conocido algoritmo NSGA-II (Fast Non-dominated Sorting Genetic Algorithm) [DPA+02] a este problema: MODENA (Multi-Objective DEsign of Nucleic Acids). Este enfoque multiobjetivo explora el conjunto aproximado de soluciones óptimas de Pareto no-dominadas en el espacio objetivo de dos funciones: (i) estabilidad y (ii) similitud de la estructura. En [Tan12], el autor extiende MODENA con métodos de predicción de pseudoknots (p. ej. IPknot o HotKnots). La última versión de MODENA [Tan15] acepta múltiples estructuras secundarias objetivo.
NUPACK es un conjunto de programas para el análisis computacional de ácido nucleico que incluye un diseñador de ARN [ZWP10]. NUPACK utiliza un método similar al utilizado por RNA-SSD, sin embargo el objetivo de NUPACK es encontrar una secuencia de ARN que minimice el defecto de ensamblaje, en lugar de la energía de la estructura (MFE).
En [LAS+12], los autores presentan un algoritmo genético para el diseño de secuencias de ARN: fRNAkenstein. En comparación con otras técnicas evolutivas (p. ej. MODENA), la principal desventaja de fRNAkenstein es que fue escrito en un lenguaje de programación interpretado (Python), lo que conlleva un tiempo de ejecución prohibitivo a medida que aumenta la longitud de la estructura objetivo.
Matthies et al. [MBT12] desarrollan el algoritmo Dynamics in Sequence Space Optimization (DSS-Opt). DSS-Opt utiliza múltiples funciones como indicadores de calidad de secuencia: (i) nearest-neighbor model energies, (ii) diseño negativo, (iii) heterogeneidad de la secuencia y (iv) restricción de composición.
RNAiFOLD es un algoritmo basado en programación de restricciones y desarrollado por J. A. García-Martín et al. [GCD13], [GDC15]. Permite al usuario especificar uno de tres criterios de optimización diferentes: (i) MFE, (ii) energía libre o (iii) defecto del conjunto. Además, proporciona una amplia variedad de restricciones de diseño, como límites de pares de bases, motifs específicos, etc.
EteRNA [LKL+14] es un juego online de ciencia ciudadana dedicado a resolver el problema de plegamiento inverso de ARN. El proyecto se llevó a cabo en 2014 y contó con una comunidad de 37,000 no-expertos dedicados a resolver diferentes estructuras objetivo que posteriormente eran probadas en laboratorios reales con el fin de aprender nuevas reglas de diseño que mejoraran sustancialmente la precisión experimental de los algoritmos. A partir de este proyecto, los autores presentan Eterna100 [AFK+16], un conjunto estándar de estructuras que se utilizan para desafiar y evaluar las nuevas generaciones de algoritmos de diseño de ARN.
En [RVC+19] se presenta una metaheurística multiobjetivo para el diseño de RNA, denominado m2dRNAs (Multiobjective Metaheuristic To Design RNA Sequences). En este trabajo, todas las secuencias de ARN reportadas deben plegarse de forma idéntica a la estructura de entrada (restricción de similitud), optimizando simultáneamente tres funciones objetivo: (i) función de partición (energía libre del conjunto), (ii) diversidad del conjunto y (iii) composición de nucleótidos.
Una vez repasados los antecedentes y estado actual del problema, vemos que se ha abordado desde diferentes puntos de vista, pero la mayoría mediante el uso de heurísticas clásicas. Por lo tanto, el uso de nuevas tendencias de inteligencia artificial supondría la consecución de avances muy importantes para este problema biológico. Por otro lado, prácticamente ninguna de las aplicaciones propuestas hacen uso de técnicas paralelas en entornos de altas prestaciones, por lo que encontramos otro reto importante: la aplicación de nuevas tecnologías y niveles de paralelismo en el diseño de secuencias de ARN. Podemos concluir, por tanto, que la combinación de novedosas técnicas de inteligencia artificial con computación de altas prestaciones es, sin lugar a dudas, una muy prometedora línea de investigación a explotar para cualquier problema de Biología computacional, y en especial para el diseño de secuencias de ARN.
|
[AFH+04] |
Andronescu, M., Fejes, A. P., Hutter, F., Hoos, H. H., & Condon, A. (2004). A New Algorithm for RNA Secondary Structure Design. Journal of Molecular Biology, 336(3), 607-624. https://doi.org/10.1016/j.jmb.2003.12.041 |
|
[AFK+16] |
Anderson-Lee, J., Fisker, E., Kosaraju, V., Wu, M., Kong, J., Lee, J., Lee, M., Zada, M., Treuille, A., & Das, R. (2016). Principles for Predicting RNA Secondary Structure Design Difficulty. Journal of Molecular Biology, 428(5), 748-757. https://doi.org/10.1016/j.jmb.2015.11.013 |
|
[AHC07] |
Aguirre-Hernández, R., Hoos, H. H., & Condon, A. (2007). Computational RNA secondary structure design: empirical complexity and improved methods. BMC Bioinformatics, 8(1), 34. https://doi.org/10.1186/1471-2105-8-34 |
|
[BB06] |
Busch, A., & Backofen, R. (2006). INFO-RNA -- A fast approach to inverse RNA folding. Bioinformatics, 22(15), 1823-1831. https://doi.org/10.1093/bioinformatics/btl194 |
|
[BRS20] |
Bonnet, É., Rzążewski, P., & Sikora, F. (2020). Designing RNA Secondary Structures Is Hard. Journal of Computational Biology, 27(3), 302-316. https://doi.org/10.1089/cmb.2019.0420 |
|
[Cec19] |
Cecilia, J. M. (2019). Guest editors’ note: Special issue on novel high-performance computing algorithms and platforms in bioinformatics. The International Journal of High Performance Computing Applications, 34(1), 3-4. https://doi.org/10.1177/1094342019889705 |
|
[CHF20] |
Chicco, D., Heider, D., & Facchiano, A. (2020). Editorial: Artificial Intelligence Bioinformatics: Development and Application of Tools for Omics and Inter-Omics Studies. Frontiers in Genetics, 11. https://doi.org/10.3389/fgene.2020.00309 |
|
[DPA+02] |
Deb, K., Pratap, A., Agarwal, S., & Meyarivan, T. (2002). A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Transactions on Evolutionary Computation, 6(2), 182-197. https://doi.org/10.1109/4235.996017 |
|
[GCD13] |
García-Martín, J. A., Clote, P., & Dotu, I. (2013). RNAiFOLD: A constraint programming algorithm for RNA inverse folding and molecular design. Journal of Bioinformatics and Computational Biology, 11(2), 1350001. https://doi.org/10.1142/s0219720013500017 |
|
[GDC15] |
García-Martin, J. A., Dotu, I., & Clote, P. (2015). RNAiFold 2.0: a web server and software to design custom and Rfam-based RNA molecules. Nucleic Acids Research, 43(W1), W513-W521. https://doi.org/10.1093/nar/gkv460 |
|
[GLB+08] |
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neubock, R., & Hofacker, I. L. (2008). The Vienna RNA Websuite. Nucleic Acids Research, 36(Web Server), W70-W74. https://doi.org/10.1093/nar/gkn188 |
|
[GSA+16] |
Greenbury, S. F., Schaper, S., Ahnert, S. E., & Louis, A. A. (2016). Genetic Correlations Greatly Increase Mutational Robustness and Can Both Reduce and Enhance Evolvability. PLOS Computational Biology, 12(3), e1004773. https://doi.org/10.1371/journal.pcbi.1004773 |
|
[HFS+94] |
Hofacker, I. L., Fontana, W., Stadler, P. F., Bonhoeffer, L. S., Tacker, M., & Schuster, P. (1994). Fast folding and comparison of RNA secondary structures. Monatshefte Für Chemie/Chemical Monthly, 125(2), 167-188. https://doi.org/10.1007/bf00818163 |
|
[LAS+12] |
Lyngsø, R. B., Anderson, J. W., Sizikova, E., Badugu, A., Hyland, T., & Hein, J. (2012). Frnakenstein: multiple target inverse RNA folding. BMC Bioinformatics, 13(1), 260. https://doi.org/10.1186/1471-2105-13-260 |
|
[LKL+14] |
Lee, J., Kladwang, W., Lee, M., Cantu, D., Azizyan, M., Kim, H., Limpaecher, A., Gaikwad, S., Yoon, S., Treuille, A., & Das, R. (2014). RNA design rules from a massive open laboratory. Proceedings of the National Academy of Sciences, 111(6), 2122-2127. https://doi.org/10.1073/pnas.1313039111 |
|
[MBT12] |
Matthies, M. C., Bienert, S., & Torda, A. E. (2012). Dynamics in Sequence Space for RNA Secondary Structure Design. Journal of Chemical Theory and Computation, 8(10), 3663-3670. https://doi.org/10.1021/ct300267j |
|
[RVC+19] |
Rubio-Largo, A., Vanneschi, L., Castelli, M., & Vega-Rodríguez, M. A. (2019). Multiobjective Metaheuristic to Design RNA Sequences. IEEE Transactions on Evolutionary Computation, 23(1), 156-169. https://doi.org/10.1109/tevc.2018.2844116 |
|
[Tan10] |
Taneda, A. (2010). MODENA: a multi-objective RNA inverse folding. Advances and Applications in Bioinformatics and Chemistry, 1. https://doi.org/10.2147/aabc.s14335 |
|
[Tan12] |
Taneda, A. (2012). Multi-Objective Genetic Algorithm for Pseudoknotted RNA Sequence Design. Frontiers in Genetics, 3. https://doi.org/10.3389/fgene.2012.00036 |
|
[Tan15] |
Taneda, A. (2015). Multi-objective optimization for RNA design with multiple target secondary structures. BMC Bioinformatics, 16(1). https://doi.org/10.1186/s12859-015-0706-x |
|
[ZPV+13] |
Zhou, Y., Ponty, Y., Vialette, S., Waldispühl, J., Zhang, Y. and Denise, A. (2013). Flexible RNA design under structure and sequence constraints using formal languages. Proc. ACM-BCB - ACM Conference on Bioinformatics, Computational Biology and Biomedical Informatics. hal-00823279, version 2. https://hal.inria.fr/hal-00823279 |
|
[ZWP10] |
Zadeh, J. N., Wolfe, B. R., & Pierce, N. A. (2010). Nucleic acid sequence design via efficient ensemble defect optimization. Journal of Computational Chemistry, 32(3), 439-452. https://doi.org/10.1002/jcc.21633 |

Investigador Principal (arl@unex.es)

Investigador (jangomez@unex.es)

Investigador (granado@unex.es)

Investigador (sesaji@unex.es)